
Conţinut
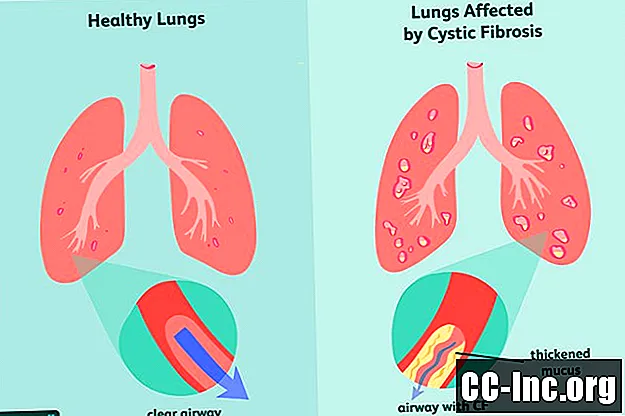
Fibroza chistică (CF) este o tulburare moștenită, care pune viața în pericol, care dăunează plămânilor și tractului digestiv. Este cauzată de o genă defectă care declanșează producerea de mucus îngroșat care înfundă căile respiratorii și blochează secreția enzimelor digestive.Simptomele sunt progresive și adesea severe și pot include probleme de respirație, infecții pulmonare recurente, creștere slabă, infertilitate masculină și inflamația cronică a pancreasului, ficatului, rinichilor și inimii.
CF poate fi diagnosticat cu teste de sânge, screening genetic și o procedură cunoscută sub numele de test de clorură de sudoare.
Deși nu există nici un remediu pentru CF, există tratamente care pot îmbunătăți atât durata cât și calitatea vieții.
Acestea includ tehnici de eliminare a căilor respiratorii, antibiotice inhalate, diluanți de mucus, enzime pancreatice, o dietă bogată în calorii și medicamente de generație mai nouă cunoscute sub numele de modulatori CFTR. În cazurile severe, poate fi necesar un transplant pulmonar.
Simptome ale fibrozei chistice
Ca o tulburare genetică, fibroza chistică este ceva cu care te naști. Poate prezenta sau nu simptome la momentul nașterii și poate dura adesea luni sau chiar ani înainte ca orice semn de boală să apară. Până în acel moment, plămânii și tractul digestiv ar fi putut suferi deja daune care nu pot fi anulate.
Cele mai frecvente semne și simptome timpurii ale CF includ:
- Blocarea primului scaun al bebelușului (meconiu)
- Piele cu gust sărat
- O tuse cronică, respirație șuierătoare sau spută colorată
- Scaune libere, grase și de obicei cu miros urât
- Infecție pulmonară, adesea recurentă
- Creșterea slabă și eșecul de a prospera
Cu excepția cazului în care aceste simptome pot fi controlate, stresul asupra plămânilor (și incapacitatea de a crește în greutate) pot avea un efect cumulativ, afectând mai multe organe și crescând riscul de complicații ale bolii.
Unele dintre complicațiile mai caracteristice includ:
- Pubertate întârziată
- Bronșiectazii (îngroșarea cronică a pereților plămânilor)
- Pierdere în greutate
- Pancreatită (inflamație a pancreasului)
- Infertilitatea masculină
- Hipertensiune pulmonară (hipertensiune arterială în plămâni)
- Pietre biliare
- Diabet zaharat legat de fibroza chistică
- Cor pulmonale (insuficiență cardiacă dreaptă)
- Ciroza (cicatrici funcționale ale ficatului)
Deoarece CF provoacă leziuni progresive ale celulelor și țesuturilor, orice deteriorare a plămânilor și a altor organe va fi în mare parte ireversibilă. Moartea va fi cel mai adesea rezultatul insuficienței respiratorii, urmată de insuficiența cardiacă și insuficiența hepatică.
Simptomele fibrozei chistice
Cauze
Fibroza chistică este cauzată de mutația genei receptorului transmembranar al fibrozei chistice (CFTR), care este responsabilă de producerea proteinei CFTR. Aceasta este proteina de care organismul are nevoie pentru a regla fluxul de sare și apă în și din celule . Dacă proteina este deformată sau defectă, aceasta poate provoca deshidratare la suprafața unei celule, ducând la îngroșarea mucusului din jur.
CF este o tulburare autosomală recesivă, ceea ce înseamnă că trebuie să moșteniți mutația CFTR atât de la mama, cât și de la tată, pentru a avea boala. Dacă moșteniți doar o genă defectă, nu veți avea CF, dar în schimb veți fi purtătorul genei mutante.
Puteți moșteni boala dacă fiecare dintre părinții dvs. are fie o mutație CFTR, fie CF în sine. Dacă ambii părinți sunt purtători, ați avea:
- 25% șanse de a avea CF
- 50% șanse de a fi transportator
- 25% șanse de a fi neafectat
Pe de altă parte, dacă unul dintre părinții dvs. este purtător și celălalt are CF, aveți șanse de 50/50 fie să aveți CF, fie să fiți purtător.
Fibroza chistică este una dintre cele mai frecvente boli genetice, afectând aproximativ unul din 2.500 de copii născuți în Statele Unite.
Este cel mai frecvent printre caucazieni și hispanici și apare mai rar la persoanele de origine africană sau asiatică.
Factorii de risc ai fibrozei chisticeDiagnostic
Există câteva teste utilizate pentru diagnosticarea fibrozei chistice. Acestea funcționează fie prin detectarea directă a mutației CFTR, fie prin măsurarea indirectă a modificărilor biologice în concordanță cu boala. Metoda de diagnostic poate varia în timpul sarcinii, când se naște copilul sau oricând după aceea.
Ghid de discuții despre medicul pentru fibroză chistică
Obțineți ghidul nostru imprimabil pentru următoarea programare a medicului pentru a vă ajuta să puneți întrebările corecte.

Dintre cele două teste standard utilizate în mod obișnuit pentru diagnosticarea CF:
- Testarea clorurii sudoripare, cunoscut și sub numele de testul transpirației, măsoară cantitatea de clorură de pe piele. Deoarece CF interferează cu transferul de sare către și din celule, va exista o acumulare de sare în transpirație.
- Testarea genetică CFTR este folosit pentru a detecta cele mai frecvente mutații ale mutației CFTR. În timp ce există peste 2.000 de mutații CFTR despre care se știe că provoacă fibroză chistică, cele 23 incluse în panoul standard reprezintă cei mai probabili suspecți.
În timpul sarcinii, testul genetic CFTR poate fi utilizat pentru a testa fluidele obținute printr-o amniocenteză sau celule extrase prin eșantionarea villusului corionic (CVS).
Screening pentru nou-născuți este, de asemenea, utilizat în mod standard pentru a diagnostica CF și este astăzi mandatat în toate cele 50 de state și districtul Columbia. Ceea ce presupune acest lucru va diferi în funcție de locul în care locuiți în Statele Unite. Dacă rezultatele screeningului nou-născutului sunt pozitive, un test de transpirație ar fi folosit pentru a confirma diagnosticul.
Cum este diagnosticată fibroza chisticăTratament
Deși nu există un remediu pentru fibroza chistică, progresele în tratament au extins durata de viață a celor care trăiesc cu boala.
Scopul tratamentului cu CF este de patru ori: prevenirea infecțiilor, menținerea funcției pulmonare, normalizarea digestiei și încetinirea progresiei bolii.
Printre instrumentele terapeutice utilizate pentru gestionarea CF:
- Tehnici de eliminare a căilor aeriene (ACT) sunt efectuate pentru a disloca și a expulza mucusul acumulat din plămâni. Tehnicile includ tuse cu puf, percuție toracică sau oscilația peretelui toracic.
- O dietă bogată în grăsimi, bogată în calorii este utilizat pentru a compensa malabsorbția grăsimilor, proteinelor și nutrienților din intestine.
- Suplimente de enzime pancreatice sunt folosite pentru a întări enzimele digestive pe care pancreasul nu le poate produce din cauza acumulării excesive de mucus.
- Antibiotice sunt luate zilnic pentru a preveni infecțiile pulmonare bacteriene.
- Mucolitice-medicamente utilizate pentru subțierea mucusului înainte de ACT-pot fi utilizate.
- Modulatori CFTR sunt o nouă clasă de medicamente care pot corecta anumite defecte ale proteinei CFTR și le pot restabili funcția de reglare.
- Terapia cu oxigen poate fi utilizat în timpul episoadelor acute când respirația dumneavoastră a fost grav afectată.
- Nutriție enterală, cunoscut și sub numele de hrănire cu tub, poate fi utilizat dacă nu puteți susține greutatea printr-o nutriție normală.
- Transplantul pulmonar se ia în considerare atunci când plămânii nu mai pot susține supraviețuirea fără ventilație mecanică.
Copiind
În 1938, când fibroza chistică a fost clasificată pentru prima dată ca boală, copiii au trăit rar după primul lor an de viață. Până în anii 1980, se putea aștepta să trăiască până la 20-25 de ani. Astăzi, imaginea s-a schimbat în întregime cu persoanele care trăiesc bine la 40 de ani și chiar la 50 de ani, dacă tratamentul este început devreme și respectat.
Acest lucru nu înseamnă că CF este mai puțin grav decât a fost vreodată. Este un eveniment care schimbă viața, necesitând diligență și consistență pentru a face față nu numai bolii, ci și pentru a trăi cel mai înalt nivel de viață posibil.
În acest scop, trebuie să normalizați CF în viața dvs. prin stabilirea rutinelor și practicilor pentru a evita urcușurile și coborâșurile care pot provoca stres și pot crește handicapul. Printre considerații, ar trebui să:
- Gestionează-ți nutriția. Persoanele cu FC au nevoie adesea de două ori mai multe calorii zilnice decât alte persoane.
- Fă sport regulat. Rutinele de fitness ar trebui să implice în mod ideal un minim de 20 până la 30 de minute de activitate aerobă de trei ori pe săptămână. Găsiți ceva plăcut pe care îl puteți face o viață întreagă.
- Păstrați-vă bine hidratat. Acest lucru face ca plămânii și intestinele să funcționeze corect. În funcție de vârstă, ar trebui să beți nu mai puțin de șase până la opt pahare înalte de apă pe zi.
- Efectuați corect distanța căilor respiratorii. Pe măsură ce nevoile dvs. de sănătate se schimbă, la fel se pot schimba și tipurile de instrumente de eliminare de care aveți nevoie. Discutați cu pneumologul sau terapeutul fizic dacă nu obțineți rezultatele pe care ar trebui.
- Căutați sprijin. În plus față de prieteni și familie, puteți contacta cel mai apropiat capitol al Fundației pentru Fibroză Chistică (CFF) pentru a vă conecta la o rețea de asistență din zona dvs.
- Căutați ajutor financiar. CFF oferă servicii care ajută familiile să facă față mai bine costului ridicat al tratamentului cu FC.
Un cuvânt de la Verywell
În timp ce screening-ul nou-născuților a crescut dramatic rata diagnosticelor de FC la bebeluși, peste 25% din diagnostice se fac doar în anii copilăriei, adolescenței și primilor adulți.
Acest lucru este problematic, deoarece diagnosticul și tratamentul timpuriu pot preveni multe dintre complicațiile mai severe ale CF înainte de a se putea produce vătămări grave. În timp ce tratamentul nu poate opri sau inversa boala, acesta poate asigura încă mulți ani fără boală.
În acest scop, este important să cunoașteți simptomele timpurii ale CF și să discutați cu medicul dumneavoastră dacă bănuiți că copilul dumneavoastră poate avea boala. Acest lucru este valabil mai ales în statele care analizează numai testele de sânge IRT, ceea ce ar putea duce la un procent de 5% dintre copii care suferă fie un diagnostic întârziat, fie un rezultat fals negativ, potrivit cercetărilor de la Școala de Medicină și Sănătate Publică a Universității din Wisconsin. .
Ce simptome vă puteți aștepta cu fibroza chistică?