
Conţinut
- cauze
- Simptome
- Examene și teste
- Tratament
- Grupuri de suport
- Perspectiva (prognoza)
- Când să vă adresați unui specialist medical
- profilaxie
- Nume alternative
- Imagini
- Referințe
- Data de examinare 10/15/2018
Boala Niemann-Pick (NPD) este un grup de boli transmise prin familii (moștenite) în care substanțele grase numite lipide se colectează în celulele splinei, ficatului și creierului.
Există trei forme comune ale bolii:
- Tipul A
- Tipul B
- Tip C
Fiecare tip implică diferite organe. Poate sau nu poate implica sistemul nervos și respirația. Fiecare poate provoca simptome diferite și poate să apară la diferite momente de-a lungul vieții.
cauze
Tipurile NPD A și B apar atunci când celulele din organism nu au o enzimă numită sfingomielinază acidă (ASM). Această substanță ajută la descompunerea (metabolizarea) unei substanțe grase numită sfingomielină, care se găsește în fiecare celulă din organism.
Dacă ASM lipsește sau nu funcționează corect, sfingomielina se acumulează în interiorul celulelor. Acest lucru vă ucide celulele și face greu organele să funcționeze corect.
Tipul A apare în toate rasele și etnicii. Este mai frecventă în populația evreiască din Ashkenazi (est-europeană).
Tipul C apare atunci când organismul nu poate descompune în mod corespunzător colesterolul și alte grăsimi (lipide). Acest lucru duce la prea mult colesterol în ficat și splină și prea mult alte lipide din creier. Tipul C este cel mai des întâlnit printre Puerto Ricans de origine spaniolă.
Tipul C1 este o varianta de tip C. Acesta implica un defect care interfereaza cu modul in care colesterolul se misca intre celulele creierului. Acest tip a fost văzut doar în francezii canadieni din județul Yarmouth, Nova Scotia.
Simptome
Simptomele variază. Alte afecțiuni de sănătate pot provoca simptome similare. Etapele timpurii ale bolii pot produce doar câteva simptome. O persoană nu poate avea niciodată toate simptomele.
Tipul A începe, de obicei, în primele câteva luni de viață. Simptomele pot include:
- Abdomen (zona abdomenului) umflată în decurs de 3 până la 6 luni
- Cocos roșu cireș la partea din spate a ochiului (pe retină)
- Dificultăți de alimentare
- Pierderea abilităților motorii timpurii (se agravează în timp)
Simptomele tip B sunt de obicei mai ușoare. Ele apar în copilăria târzie sau în adolescență. Tumorile abdominale pot apărea la copiii mici. Nu există aproape nici o implicare a creierului și a sistemului nervos, cum ar fi pierderea abilităților motorii. Unii copii pot avea infecții respiratorii repetate.
Tipurile C și C1 afectează în mod obișnuit copiii de vârstă școlară. Cu toate acestea, aceasta poate apărea oricând între copilăria timpurie până la maturitate. Simptomele pot include:
- Dificultate în mișcare a membrelor, care pot duce la mers instabil, agitate, probleme de mers pe jos
- Splină mărită
- Ficat mărit
- Icter la naștere (sau la scurt timp după)
- Dificultăți de învățare și declin intelectual
- convulsii
- Vorbire neclară, neregulată
- Pierderea bruscă a tonusului muscular care poate duce la căderi
- tremors
- Probleme în mișcarea ochilor în sus și în jos
Examene și teste
Un test de sânge sau de măduvă osoasă poate fi efectuat pentru a diagnostica tipurile A și B. Testul poate indica cine are boala, dar nu arată dacă sunteți transportator. Testele ADN pot fi efectuate pentru diagnosticarea purtătorilor de tip A și B.
O biopsie cutanată este de obicei făcută pentru a diagnostica tipurile C și D. Furnizorul de îngrijire medicală urmărește modul în care celulele pielii cresc, se mută și stochează colesterolul. Testele ADN pot fi, de asemenea, efectuate pentru a căuta cele două gene care provoacă acest tip de boală.
Alte teste pot include:
- Mărirea măduvei osoase
- Biopsia hepatică (de obicei nu este necesară)
- Examenul de ochi cu lampă
- Teste pentru a verifica nivelul ASM
Tratament
În acest moment nu există un tratament eficient pentru tipul A.
Transplanturile de măduvă osoasă pot fi încercate pentru tipul B. Cercetătorii continuă să studieze tratamente posibile, inclusiv înlocuirea enzimelor și terapia genică.
Un nou medicament numit miglustat este disponibil pentru simptomele sistemului nervos de tip C.
Colesterolul ridicat poate fi administrat cu o dietă sănătosă, cu un nivel redus de colesterol sau cu medicamente. Cu toate acestea, cercetarea nu arata ca aceste metode opresc boala de la a se inrautati sau de a schimba modul in care celulele descompun colesterolul. Medicamentele sunt disponibile pentru controlul sau ameliorarea multor simptome, cum ar fi pierderea bruscă a tonusului muscular și convulsii.
Grupuri de suport
Aceste organizații pot oferi sprijin și mai multe informații despre boala Niemann-Pick:
- Institutul Național de Tulburări neurologice și accident vascular cerebral - www.ninds.nih.gov/Disorders/All-Disorders/Niemann-Pick-Disease-Information-Page
- Fundația Națională Niemann-Pick Disease - nnpdf.org
- Organizația Națională pentru Tulburări Rare - rarediseases.org/rare-diseases/niemann-pick-disease-type-c
Perspectiva (prognoza)
NPD tip A este o boală severă. De obicei, duce la deces până la vârsta de 2 sau 3 ani.
Cei cu tip B pot trăi în copilăria târzie sau la maturitate.
Un copil care prezintă semne de tip C înainte de vârsta de 1 ani nu poate trăi până la vârsta școlară. Cei care prezintă simptome după ce au intrat în școală pot să trăiască în adolescența lor de la mijlocul până la sfârșitul vieții. Unii pot trăi în anii '20.
Când să vă adresați unui specialist medical
Faceți o întâlnire cu furnizorul dvs. dacă aveți un istoric familial de boală Niemann-Pick și intenționați să aveți copii. Se recomandă consilierea și screening-ul genetic.
Apelați-vă furnizorul dacă copilul are simptome ale acestei boli, inclusiv:
- Probleme de dezvoltare
- Probleme legate de hrănire
- Scădere în greutate scăzută
profilaxie
Toate tipurile de Niemann-Pick sunt autosomale recesive. Acest lucru înseamnă că ambii părinți sunt purtători. Fiecare părinte are o copie a genei anormale fără a avea semne ale bolii.
Când ambii părinți sunt purtători, există o șansă de 25% ca copilul lor să aibă boala și o șansă de 50% ca copilul să fie transportator.
Testarea detectării transportatorului este posibilă numai dacă este identificat defectul genetic. Defectele implicate în tipurile A și B au fost bine studiate. Testele ADN pentru aceste forme de Niemann-Pick sunt disponibile.
Defectele genetice au fost identificate în ADN-ul multor persoane cu tip C. Este posibil să se poată diagnostica persoanele care poartă gena anormală.
Câteva centre oferă teste pentru a diagnostica un copil încă în uter.
Nume alternative
NPD; Deficit de sphingomyelinase; Tulburarea de stocare a lipidelor - boala Niemann-Pick; Boala de stocare a lizozomilor - Niemann-Pick
Imagini
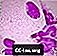
Celulele spumoase Niemann-Pick
Referințe
Kliegman RM, Stanton BF, St. Geme JW, Schor NF. Defectele metabolismului lipidelor. În: Kliegman RM, Stanton BF, St. Geme JW, Schor NF, eds. Nelson Manual de Pediatrie. 20 ed. Philadelphia, PA: Elsevier; 2016: cap 86.
Patterson MC, Hendriksz CJ; Grupul de lucru pentru linii directoare NP-C, et al. Recomandări pentru diagnosticarea și managementul bolii Niemann-Pick tip C: o actualizare. Mol Genet Metab. 2012; 106 (3): 330-344. PMID: 22572546 www.ncbi.nlm.nih.gov/pubmed/22572546.
Turnpenny PD, Ellard S. Erori inbornate ale metabolismului. In: Turnpenny PD, Ellard S, eds. Elementele Emery de Genetică Medicală. 15 ed. Philadelphia, PA: Elsevier; 2017: cap 18.
Data de examinare 10/15/2018
Actualizat de: Anna C. Edens Hurst, MD, MS, asistent universitar în genetica medicală, Universitatea din Alabama la Birmingham, Birmingham, AL. Revizuire oferită de VeriMed Healthcare Network. De asemenea, revizuit de către David Zieve, MD, MHA, Director Medical, Brenda Conaway, Director Editorial, și A.D.A.M. Echipa editorială.