
Conţinut
- cauze
- Simptome
- Examene și teste
- Tratament
- Grupuri de suport
- Perspectiva (prognoza)
- Posibile complicații
- Când să vă adresați unui specialist medical
- profilaxie
- Nume alternative
- Instrucțiuni pentru pacient
- Imagini
- Referințe
- Data de examinare 2/19/2018
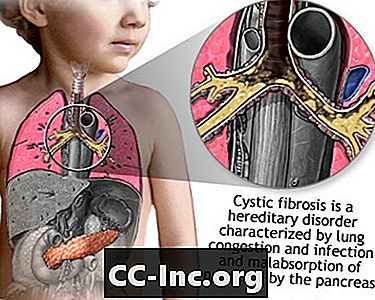
Fibroza chistică este o boală care provoacă mucus gros, lipicios, care se acumulează în plămâni, în tractul digestiv și în alte zone ale corpului. Este una dintre cele mai frecvente boli pulmonare cronice la copii și adulți tineri. Este o afecțiune care pune viața în pericol.
cauze
Fibroza chistică (CF) este o boală care este transmisă prin intermediul familiilor. Este cauzată de o gena defectă care face corpul să producă un lichid anormal de gros și lipicios, numit mucus. Acest mucus se acumulează în căile respiratorii ale plămânilor și în pancreas.
Acumularea de mucus are ca rezultat infecții pulmonare amenințătoare de viață și probleme digestive grave. Boala poate afecta, de asemenea, glandele sudoripare și sistemul de reproducere al bărbatului.
Mulți oameni poartă o gena CF, dar nu au simptome. Acest lucru se datorează faptului că o persoană cu CF trebuie să moștenească 2 gene defecte, 1 de la fiecare părinte. Unii americani albi au gena CF. Este mai des întâlnită printre cei de origine nordică sau centrală europeană.
Majoritatea copiilor cu CF sunt diagnosticați după vârsta de 2 ani. Pentru un număr mic, boala nu este detectată până la vârsta de 18 ani sau mai mult. Acești copii au adesea o formă mai ușoară a bolii.
Simptome
Simptomele la nou-născuți pot include:
- Creștere întârziată
- Eșecul în greutate în mod normal în timpul copilăriei
- Nu există mișcări intestinale în primele 24 până la 48 de ore de viață
- Piele degustată cu sărat
Simptomele legate de funcția intestinului pot include:
- Durerea de burtă din constipație severă
- Boală crescută, balonare sau burtă, care apare umflată
- Greață și pierderea apetitului
- Scaune care sunt de culoare palidă sau de argilă, mirosuri murdare, au mucus sau care plutesc
- Pierdere în greutate
Simptomele legate de plămâni și sinusuri pot include:
- Tuse sau creșterea mucusului în sinusuri sau plămâni
- Oboseală
- Congestie nazală cauzată de polipi nazali
- Episoadele de pneumonie repetate (simptomele pneumoniei la cineva cu fibroză chistică includ febră, tuse crescută și dificultăți de respirație, mucus crescut și pierderea poftei de mâncare)
- Dureri sau presiuni cauzate de infecții sau polipi
Simptome care pot fi observate mai târziu în viață:
- Infertilitatea (la bărbați)
- Inflamația repetată a pancreasului (pancreatită)
- Simptome respiratorii
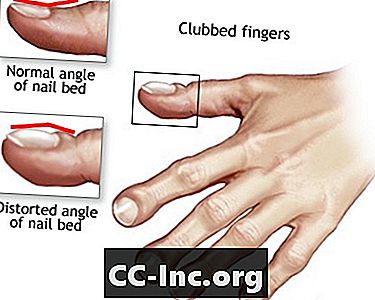
- Degetele cu degete
Examene și teste
Un test de sânge se face pentru a ajuta la detectarea CF. Testul caută schimbări în gena CF. Alte teste utilizate pentru a diagnostica CF includ:
- Testul tripsinogen imunoreactiv (IRT) este un test standard de screening pentru nou-născut pentru CF. Un nivel ridicat de IRT sugerează posibil CF și necesită teste suplimentare.
- Testul de clorură de transpirație este testul standard de diagnosticare pentru CF. Un nivel ridicat de sare din sudoarea persoanei este un semn al bolii.
Alte teste care identifică problemele care pot fi legate de CF includ:
- Chist raze X sau scanare CT
- Fecal test de grăsime
- Testul funcției pulmonare
- Măsurarea funcției pancreatice
- Testul de stimulare secretină
- Trypsin și chymotrypsin în scaun
- GI superioară și serii mici de intestin
Tratament
Un diagnostic precoce al planului de tratament și al tratamentului poate îmbunătăți atât supraviețuirea, cât și calitatea vieții. Urmărirea și monitorizarea sunt foarte importante. Atunci când este posibil, ar trebui să se acorde îngrijire la o clinică de specialitate pentru fibroza chistică. Când copiii ajung la vârsta adultă, aceștia trebuie să se transfere la un centru de specialitate pentru fibroza chistică pentru adulți.
Tratamentul pentru probleme pulmonare include:
- Antibiotice pentru prevenirea și tratarea infecțiilor pulmonare și sinusale. Acestea pot fi administrate pe cale orală sau administrate în vene sau prin tratamente de respirație. Persoanele cu CF pot lua antibiotice doar atunci când este necesar sau tot timpul. Dozele sunt adesea mai mari decât în mod normal.
- Medicamente inhalate care ajută la deschiderea căilor respiratorii.
- Alte medicamente care sunt date de un tratament de respirație pentru mucus subțire și care ușurează tusea sunt enzima DNAse. terapie și soluții de sare foarte concentrate (soluție salină hipertonică).
- Vaccinul împotriva gripei și vaccinul polizaharidic pneumococ (PPV) anual (întrebați-vă furnizorul de servicii medicale).
- Transplantul pulmonar este o opțiune în unele cazuri.
- Tratamentul cu oxigen poate fi necesar deoarece boala pulmonară se agravează.
Problemele pulmonare sunt, de asemenea, tratate cu terapii pentru a subțiri mucusul. Acest lucru face mai ușoară tuserea mucusului din plămâni.
Aceste metode includ:
- Activitate sau exercițiu care vă determină să respirați adânc
- Dispozitivele utilizate în timpul zilei pentru a ajuta la curățarea căilor respiratorii ale mucusului prea mare
- Percuție toracică manuală (sau fizioterapie în piept), în care un membru al familiei sau un terapeut bate ușor pieptul persoanei, spatele și zona sub brațe
Tratamentul pentru probleme intestinale și nutriționale poate include:
- O dietă specială bogată în proteine și calorii pentru copii și adulți mai mari
- Pancreatice enzime pentru a ajuta la absorbția de grăsimi și proteine, care sunt luate cu fiecare masă
- Suplimentele de vitamine, în special vitaminele A, D, E și K
- Furnizorul dvs. vă poate sfătui alte tratamente dacă aveți scaune foarte dure
Ivacaftor și Lumacaftor sunt medicamente care tratează anumite tipuri de CF. Îmbunătățește funcția uneia dintre genele defecte care cauzează CF. Ca urmare, există o cantitate mai mică de mucus gros în plămâni. Alte simptome de CF sunt, de asemenea, îmbunătățite.
Îngrijirea și monitorizarea la domiciliu trebuie să includă:
- Evitarea fumului, a prafului, a murdăriei, a vaporilor, a produselor chimice de uz casnic, a fumului pentru șemineu și a mucegaiului sau mucegaiului.
- Oferă o mulțime de fluide, în special la sugari și copii în condiții de temperatură ridicată, când există diaree sau scaune libere sau în timpul unei activități fizice suplimentare.
- Exercitarea de 2 sau 3 ori pe săptămână. Înotul, joggingul și ciclismul sunt opțiuni bune.
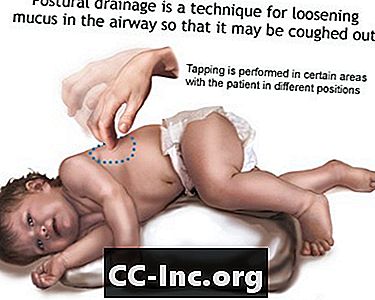
- Îndepărtarea sau aducerea de mucus sau secreții din căile respiratorii. Acest lucru trebuie făcut de la 1 la 4 ori în fiecare zi. Pacienții, familiile și îngrijitorii trebuie să învețe despre efectuarea percuțiilor toracice și a drenajului postural pentru a menține clar căile respiratorii.
Grupuri de suport
Puteți ameliora stresul bolii prin aderarea la un grup de suport pentru fibroză chistică. Împărtășirea cu alții care au experiențe și probleme comune poate ajuta familia să nu se simtă singură.
Perspectiva (prognoza)
Majoritatea copiilor cu CF trăiesc în stare bună de sănătate până când ajung la maturitate. Sunt capabili să participe la majoritatea activităților și să frecventeze școala. Mulți adulți tineri cu colegiu de terminare a CF sau găsiți locuri de muncă.
Boala pulmonară se înrăutățește până la punctul în care persoana este dezactivată. Astăzi, durata medie de viață pentru persoanele cu CF care trăiesc la vârsta adultă este de aproximativ 37 de ani.
Moartea este cel mai adesea cauzată de complicațiile pulmonare.
Posibile complicații
Cea mai obișnuită complicație este infecția cronică respiratorie.
Alte complicații includ:
- Probleme intestinale, cum ar fi calculi biliari, blocaj intestinal și prolaps rectal
- Tuse sânge
- Insuficiență respiratorie cronică
- Diabet
- infertilitate
- Boală hepatică sau insuficiență hepatică, pancreatită, ciroză biliară
- subnutriție
- Nazile nazale și sinuzita
- Osteoporoza și artrita
- Pneumonie care se întoarce
- pneumotorax
- Insuficiența cardiacă dreaptă (cor pulmonale)
Când să vă adresați unui specialist medical
Apelați-vă furnizorul dacă un copil sau copil are simptome de CF și experiențe:
- Febră, tuse crescută, modificări ale sputei sau sângelui în spută, pierderea poftei de mâncare sau alte semne de pneumonie
- Creșterea pierderii în greutate
- Mai multe mișcări frecvente ale intestinului sau scaune care sunt mirositoare sau au mai mult mucus
- Umflarea burta sau creșterea balonării
Apelați-vă furnizorul dacă o persoană cu CF dezvoltă noi simptome sau dacă simptomele se înrăutățesc, dificultăți deosebit de severe la respirație sau tuse în sânge.
profilaxie
CF nu poate fi prevenit. Screening-ul celor cu antecedente familiale ale bolii poate detecta gena CF în numeroși purtători.
Nume alternative
CF
Instrucțiuni pentru pacient
- Nutriție enterală - probleme de gestionare a copilului
- Gastrosomie tub de alimentare - bolus
- Cum sa respirati cand nu aveti suficienta respiratie
- Tub de alimentare Jejunostomy
- Drenarea posturală
Imagini
clubbing
Drenarea posturală
Degetele cu degete
Fibroză chistică
Referințe
Accurso FJ. Fibroză chistică. În: Goldman L, Schafer AI, eds. Medicina Goldman-Cecil. 25 ed. Philadelphia, PA: Elsevier Saunders; 2016: chap 89.
Egan ME, Green DM, Voynow JA. Fibroză chistică. În: Kliegman RM, Stanton BF, St. Geme JW, Schor NF, eds. Nelson Manual de Pediatrie. 20 ed. Philadelphia, PA: Elsevier; 2016: chap 403.
Farrell PM, White TB, Ren CL și colab. Diagnosticul fibrozei chistice: orientări consensuale de la Fundația pentru Fibroza Chistică. J Pediatr. 2017; 181S: S4-S15.e1. PMID: 28129811 www.ncbi.nlm.nih.gov/pubmed/28129811.
Rowe SM, Hoover W, Solomon GM, Sorscher EJ. Fibroză chistică. În: Broaddus VC, Mason RJ, Ernst JD și colab., Eds. Murray și Nadel's Manual de Medicină Respiratorie. Ed. 6 Philadelphia, PA: Elsevier Saunders; 2016: cap 47.
Taylor-Cousar JL, Munck A, McKone EF, și colab. Tezacaftor-ivacaftor la pacienții cu fibroză chistică homozigotă pentru phe508del. N Engl J Med. 2017; 377 (21): 2013-2023. PMID: 29099344 www.ncbi.nlm.nih.gov/pubmed/29099344.
Data de examinare 2/19/2018
Actualizat de: Neil K. Kaneshiro, MD, MHA, Profesor Clinic de Pediatrie, Universitatea din Washington School of Medicine, Seattle, WA. De asemenea, revizuit de către David Zieve, MD, MHA, Director Medical, Brenda Conaway, Director Editorial, și A.D.A.M. Echipa editorială.